导读碳基材料在构建可持续未来方面具有巨大潜力,但材料科学家需要工具来正确分析它们的原子结构,这决定了它们的功能特性。X射线光电子能谱(XP...
碳基材料在构建可持续未来方面具有巨大潜力,但材料科学家需要工具来正确分析它们的原子结构,这决定了它们的功能特性。X射线光电子能谱(XPS)是用于执行此操作的工具之一,但XPS结果可能难以解释。
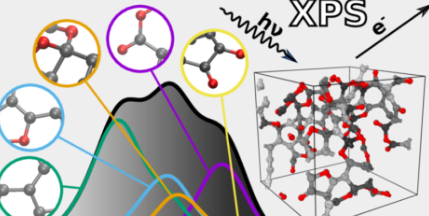
现在,Aalto的研究人员开发了一种机器学习工具来改进XPS分析,并将其作为XPS预测服务器免费提供。
XPS光谱是具有一组峰的图表,这些峰反映了构成材料的原子深处的电子的结合能。因为结合能取决于原子环境,它们可以用来推断原子在特定材料或分子中是如何连接的。然而,这也使得XPS光谱难以解释,因为许多因素会影响结合能。不同原子特征的结合能也可能重叠,使分析更加复杂。
为了解决这个问题,由MiguelCaro领导的团队开发了一种计算方法,可以基于计算机生成的结构模型预测材料的结合能谱。通过使实验观察到的结合能与计算预测相匹配,这简化了XPS数据解释。
这个想法本身并不新鲜,但问题在于准确计算材料的XPS光谱的计算难度。Caro的团队使用机器学习解决了这个问题。诀窍是训练一种廉价的计算机算法,以基于计算上廉价和昂贵的量子力学数据的有效组合来预测计算上昂贵的参考方法的结果。
计算成本较低的方法DFT不能非常准确地匹配实验结果。当一个分子有很多原子时,更准确的方法GW需要很长时间才能计算出来。“我们决定构建一个使用丰富的DFT数据的基线模型,然后使用稀缺而珍贵的GW数据对其进行改进。它奏效了!卡罗说。
由此产生的算法可以预测由碳、氢和氧组成的任何无序材料的光谱。'预测的光谱与实验获得的光谱非常接近。这为更好地整合材料的实验和计算表征打开了大门,”卡罗说。接下来,该团队计划扩展他们的技术,以包括更广泛的材料和其他类型的光谱学。